
- Unparteilichkeit
- Vertraulichkeit
- Rückführbarkeit
- Verifikation
- Rückverfolgbarkeit
Im heiligen Pentagon qualitätssicherer Analystik und Begutachtung stellt „Verifikation“ die vierte Säule. Probenahme, Probevorbereitung, Analyse und Ergebnisermittlung erfordern Schritt für Schritt und insgesamt eine Verifikation. Wie unterscheiden sich „Verifikation/Verifizierung“ im Vergleich zu „Validierung“. Der Eurachem-Guide „Terminologie bei Analytischen Messungen – Einführung in den VIM 3“ [1] definiert:
Verifizierung: „Erbringung eines objektiven Nachweises, dass eine Betrachtungseinheit die spezifizierten Anforderungen erfüllt.“
Diese Definition deckt sich mit der Begriffsbestimmung der DIN17025:2018-3. Sie ist eine „Bestätigung“, also kein Element, dass die Fähigkeit zur Anwendung einer selbstentwickelten neuen Prüfmethode kennzeichnet, sondern Verifikation / Verifizierung bestätigt zum Beispiel, dass ein validiertes möglicherweise sogar als DIN-Verfahren in einem rechtlichen Kontext vorgebenes Verfahren beherrscht wird und die vorgesehenen Ergebnisse (richtiger Messwert, vorgesehene Messunsicherheit) in Zusammenspiel mit der vorgebenen Matrix des Analyten erbringt.
Es ist nicht automatisch davon auszugehen, dass eine Prüfeinrichtung mit einem DIN-Verfahren die für ein käufliches Referenzmaterial vorgesehenen Ergebnisse misst, dass die Messbereichsgrenzen, die Messunsicherheit, die Unempfindlichkeit gegen Matrixeffekte genau wie im vorgegebenen DIN-Verfahren ausfallen. Gründe können sein: Prüfeinrichtungen verwenden selten die exakt gleichen Geräte. Die individuelle Streuung insbesondere bei Neueinführung ist zu berücksichtigen. Lösungsmittel, Chemikalien unterliegen einer Chargenvariation. Bei der Auswertung kann der zu verwendende Algorithmus durch eine novellierte Norm zu anderen Ergebnissen führen.
Die eigentliche, ausschließlich messgeräteabhängige Messung hingegen sollte aufgrund der Rückführung der Messgeräte zu keinem signifikanten Ergebnisunterschied beitragen.
Validierung: „Die Validierung ist eine Verifizierung, wobei die spezifizierten Anforderungen für den beabsichtigten Zweck angemessen sind.
Diese Definition deckt sich ebenfalls mit der Begriffsbestimmung der DIN17025:2018-3. Die „Absicht“ – also das Ziel – der Bestimmung und die „Angemessenheit“ treten zur Verifizierung. Der „Absicht“, die Dicke des Blechs eines Kotflügels zu bestimmen, ist die Verwendung eines Interferometers vermutlich nicht angemessen, obwohl es vermutlich prinzipiell möglich sein sollte. Eine Bestimmung der Kalziumkonzentration im Serum eines Patienten ist mittels Feinwaage ebenfalls möglich, aber in diesem Anwendungsfall der Gravimetrie erforderliche Probenvorbereitung stellt nicht nur einen hohen Aufwand dar, sie kann auch zu einer Messunsicherheit jenseits der Eignung für den beabsichtigten Einsatz führen.
DIN-Methoden sind in der Regel validiert.
Vor die Verwendung einer von Dritten validierten Prüfmethode stellt die analytische Korrektheit die Verifikation zum Nachweis der erforderlichen Kompetenz der Prüfeinrichtung, die diese Prüfmethode verwenden will. „Kompetenz“ erfasst in diesem Zusammenhang sowohl den technischen Bereich (Vorhandensein und Rückführung von Messgeräten, Material etc.) als auch den personellen Bereich (Fähigkeit die technischen Mittel richtig / geeignet und zielführend einzusetzen sowie die erhaltenen Zahlen zu verwertbaren Ergebnissen umzusetzen.
Was bedeutet die Forderung „Nur validierte und verifizierte Verfahren dürfen für das Ermitteln qualitätsgesicherter Ergebnissen verwendet werden“ in der Praxis?
1. Aus Normen, Richtlinien und Vorgaben entnommene Verfahren können (zunächst) als validiert gelten. Soll ein solches Verfahren außerhalb seines validierten Bereichs verwendet werden, ist es für diesen Bereich zu validieren. Ist die Bestimmung eines Parameters in einer definierten Matrix für einen bestimmten Konzentrationsbereich in einem DIN-Verfahren beschrieben (Kalziumionen in Wasser), kann dieses Verfahren nicht auf beliebig niedrige Konzentrationen oder beliebige Matrices angewandt werden. Bei der Übernahme validierter Prüfverfahren in das eigene Verfahrensportfolio unter den für die Validierung geltenden Randbedingungen genügt in der Regel eine dokumentierte Verifikation des Verfahrens.
2. Hausverfahren, also solche, die im Rahmen von Forschung und Entwicklung neu konzipiert werden, sowie die Anwendung schon validierter Verfahren außerhalb ihrer Spezifikation, also zum Beispiel mit einer geänderten Matrix, erweitertem Messbereich oder anderen Modifikationen, machen eine vollständige Validierung des Verfahrens erforderlich. Auch Versuche zur Verkürzung von Inkubationszeiten durch Temperaturanpassung, Weglassen eines Waschschritts im ELISA, eben alle Veränderungen, die geeignet sind Spezifikationen und Eignung der Validierung zu beeinflussen, erfordern eine erneute Validierung des Verfahrens.
3. Die Erfüllung der beiden vorgenannten Forderungen allein erfüllen die Ansprüche an eine qualitätssichere und verlässliche Analytik & Begutachtung einer akkreditierten Prüfeinrichtung oder einer verlässlichen / reproduzierbaren Forschung & Entwicklung noch nicht. Eine Prüfmethode erhält erst ihren Segen für die Verwendung im akkreditierten Angebot, wenn die erzielten Ergebnisse den Ergebnissen der Untersuchung des gleichen Prüfgegenstands durch andere unabhängige Prüfeinrichtungen entsprechen. Dieses Verfahren wird als „Laborvergleichsuntersuchung“ bezeichnet.
Ein oft beschrittener Weg zur Übernahme eines Verfahrens im eigenen Labor führt über zertifizierte Referenzmaterialien. Mit ihnen kann die Richtigkeit von Prüfergebnissen bei der Etablierung der neuen Prüfmethode bestätigt und die Messunsicherheit ermittelt werden. Da die Anforderungen an die Messgeräte in der mit der zugehörigen DIN, Richtlinie, den Herstellerangaben von Fertigtestsbestecken im allgemeinen festgelegt sind und die Enpunktbestimmung / Messwertaufnahme durch Überprüfung und Rückführung der Messgeräte eher in sehr geringe Maß zur Variation der Messwerte beitragen, liegt der Schwerpunkt der folgenden Betrachtung auf der Probenvorbereitung. Hier am Beispiel einer Festphasenextraktion. Neben der Probenvorbereitung trägt insbesondere die Probenahme maßgeblich zum Fehler im Endergebnis bei. Fehler in der Probenahme kann die Probenvorbereitung nicht ausgleichen. Fehler zu Beginn einer Ergebnisermittlung addieren sich, wobei die Endpunktsbestimmung den geringsten Fehlerbeitrag leistet.
Die Festphasenextraktion ist eine Methode der chemisch-analytischen Probenvorbereitung, die gelöste Analyten von der flüssigen Matrix trennt, in der sie gelöst sind.
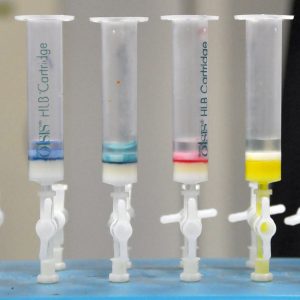
// Hier fehlen noch Links, Bilder und Informationen zur Standardmethode.
Die besondere Problematik soll am Beispiel von Endpunktsbestimmungen mittels Fotometrie veranschaulicht werden. Zur Veranschaulichung und schnellen Erfassung zeigt die obige Abbildung Farbstoffe, die in wässriger Umgebung an die Festphase (weißes Material in der Kartusche) binden.

Für die Eignung eines Analyten für eine fotometrische Bestimmung ist sein spezischen Vermögen, Licht bestimmter Wellenlängen zu absorbieren. Der obige Aufbau zeigt die Anreicherung eines Analyten (Ethinylestradiol) aus gereinigtem Klärwasser. Hier ist die Entfernung der Matrix (Wasser) die Hauptaufgabe der Festphasenextraktion. Man spricht von einer Anreicherung.
In jedem Fall muss der Analyt die Festphase unter geeigneten Bedingungen quantitativ und in einem möglichst geringen Volumen verlassen. Das Aufgefangene dieses Prozesses bezeichnet man als Eluat. In der Gesamtbilanz der Probenvorbereitung sollte idealerweise die gesamte aufgetragene Substanz im Eluat versammelt sein. Input = Output.
Die Realität sieht den Gesamtprozess jedoch sowohl verlustbehaftet als auch scheinbar analyterzeugend. Während für den ersteren Fall ein Verbleib eines Teils der Probe auf der Festphase, also eine unvollständige Elution, verantwortlich zu machen wäre, findet sich keine einfache und plausible Ereklärung für den Fall einer scheinbaren Substanzvermehrung. Wenn der letzte Gesichtspunkt unplausibel ist und mit Sicherheit den wahren Sachverehalt nicht wiedergibt, dann muss der erste Fall nicht richtiger sein, auch wenn er plausibel ist.
Das für den Auftrag verwendete Lösungsmittel (Matrix) des Analyten (Wasser, Serum, Saft, Urin …) wird durch das für das Herauslösen (Eluieren) verwendete Lösungsmittel (meist unpolarer als die Matrix) ersetzt. Alternativ und / oder ergänzend kann auch das pH-Milieu verändert werden. Wenn in einem fotometrischen Verfahren
- pH-Wert,
- Salzkonzentration,
- Proteinkonzentration,
- Art des Salzes,
- …
geändert werden, kann dies das absorbierte Lichtspektrum hoch signifikant verändern [2]. Solvatochromie, isobestischer Punkt sind aus der fotometrischen Titration bekannt [3, 4]. Ebenso kann es zu Farbänderungen in Folge von Ligandenaustausch oder Temperaturänderungen kommen [5]. Der Eiweißfehler bei Bromthymolblau ist ebenfalls bekannt [6].
Unter Solvatochromie versteht man allgemein die Beeinflussung der Farbe eines Farbstoffes durch Lösungsmittel (Zitat Wikipedia).
Das folgende Video zeigt einen solvatochromen Effekt beim Lösen von Iod in unterschiedlichen Lösungsmitteln.
Das folgende ebenfalls dem Bestand von Youtube entnommene Video demonstriert mit der Solvatofluoreszenz einen ebenfalls solvatochromen Effekt am Beispiel von Nilrot.
Noch beeindruckender die vergleichende Darstellung des in unterschiedlichen Lösungsmitteln gelosten Farbstoffs Nilrot.

Von Kuebi = Armin Kübelbeck – Eigenes Werk, CC BY-SA 3.0, Link
Nilrot bei Tageslicht (obere Reihe) und UV-Licht (366 nm, untere Reihe) in verschiedenen Lösungsmitteln. V. l. n. r.: 1. Wasser, 2. Methanol, 3. Ethanol, 4. Acetonitril, 5. Dimethylformamid, 6. Aceton, 7. Ethylacetat, 8. Dichlormethan, 9. n-Hexan, 10. tert-Butylmethylether, 11. Cyclohexan, 12. Toluol.
Die Molekularstruktur lässt aromatische Strukturen und ein ausgedehntes konjugiertes Doppelbindungssystem mit Heteroatomen erkennen.

Von Kuebi = Armin Kübelbeck – Eigenes Werk, Gemeinfrei, Link
Viele wässrig gelöste Analyten absorbieren kein sichtbares Licht. Ihre fotometrische quantitative Erfassung nutzt dann eventuell lichtabsorbierende Eigenschaften im nahen Infrarot oder im ultravioletten Bereich des Lichts.
Unter geeigneter Berücksichtigung der genannten Effekte ergibt sich demnach ein möglicherweise ganz anderes Ergebnis für die Wiederfindung. Die Würdigung möglicher Seiteneffekte ist Bestandteil der Validierung. Die Wahl eines anderen als des vorgesehenen Lösungsmittels kann signifikante Nebeneffekte zeitigen, welche die Richtigkeit der Analyse fraglich werden lassen.
Lehrvideos zur Erläuterung und der Solvatochromie sowie ihrer möglichen Anwendung
Autoren: Reichardt, Christian Jerabek, Paul Hegemann, Julian Authmann, Andreas
Lizenz: CC-Namensnennung – keine kommerzielle Nutzung – Weitergabe unter gleichen Bedingungen 3.0 Deutschland:
Sie dürfen das Werk bzw. den Inhalt zu jedem legalen und nicht-kommerziellen Zweck nutzen, verändern und in unveränderter oder veränderter Form vervielfältigen, verbreiten und öffentlich zugänglich machen, sofern Sie den Namen des Autors/Rechteinhabers in der von ihm festgelegten Weise nennen und das Werk bzw. diesen Inhalt auch in veränderter Form nur unter den Bedingungen dieser Lizenz weitergeben.
Quelle:
Anwendungen von Solvatochromie
Autoren:Reichardt, Christian Jerabek, Paul Hegemann, Julian Authmann, Andreas
Quelle: https://doi.org/10.5446/18765
Lizenz: CC-Namensnennung – keine kommerzielle Nutzung – Weitergabe unter gleichen Bedingungen 3.0 Deutschland:
Sie dürfen das Werk bzw. den Inhalt zu jedem legalen und nicht-kommerziellen Zweck nutzen, verändern und in unveränderter oder veränderter Form vervielfältigen, verbreiten und öffentlich zugänglich machen, sofern Sie den Namen des Autors/Rechteinhabers in der von ihm festgelegten Weise nennen und das Werk bzw. diesen Inhalt auch in veränderter Form nur unter den Bedingungen dieser Lizenz weitergeben.
https://www.researchgate.net/publication/259652177_Investigation_of_Pharmaceutical_Metabolites_in_Environmental_Waters_by_LC-MSMS/download
http://www.mibius.de/out/oxbaseshop/html/0/images/wysiwigpro/pH_Brosch_d%281%29.pdf
https://www.nugi-zentrum.de/fileadmin/website_uni_ulm/nugi/Experimente/Grundlagen/pH-Wert/Titration__Theorie__Indikatoren.pdf
[1] Terminologie bei Analytischen Messungen – Einführung in den VIM 3
[2] „pH-Indikatoren – Sekundenschnelle Analysen“
[3] pH-abhängige Spektren und fotometrische Titrationskurven
Bestimmung der Säurekonstanten von Bromthymolblau
Säure-Base-Indikatoren (pH-Indikatoren)
[5] Gleichgewichtsverschiebung bei Ligandenaustauschreaktionen
[6] Eiweißfehler bei Bromthymolblau